We have underestimated Covid-19 and the virus that causes it, SARS-CoV-2, at every turn. At first, we doubted that the troubling news from China was the start of a new pandemic. Then we hoped what passed for border controls would keep it from our shores. We rejected the idea of aerosols transmission for far too long. We believed that as the virus spread, it would weaken and then disappear in the summer months. With the advent of unexpectedly potent vaccines, we declared what now appears to be a premature end to the pandemic.
SARS-CoV-2 turns out to be everything we have hoped it wasn’t. A final last hope was that the virus might have a limited capacity for change. That appears not to be so. In our catalog of variants of interest and concern, we are rapidly exhausting the Greek alphabet, which now numbers variants from alpha to iota. A glimmer of optimism seemed to be that all relevant changes are to the S protein that encodes the Spike protein. If so, the reasoning went, then the repertoire of changes might be limited.
A new study by Thorne et al. throws cold water on that hypothesis. The authors find that seemingly insignificant changes in the primary nucleotide sequence have profound changes in the expression of viral genes, elevating the concentrations of some viral genes over others by almost 100 fold. The amplified genes encode proteins that suppress the cell’s ability to mount effective innate immune responses, leading to increases in the duration of viremia and transmission. The conclusion: we must analyze the entire genome for mutations that contribute to the properties of new variants, not only those within the S protein. A genomicist’s dream, a public health nightmare.
The Thorne et al. study attributes increased transmission of the Alpha (B.1.1.7) variant of SARS-CoV-2 to preferential expression of virus-encoded immune suppression genes and proteins, in addition to the changes in the spike protein. Paradoxically, early infection by SARS-CoV-2 suppresses the early immune response before inciting hyper-immune responses later. Early suppression of the innate immune system accounts for the ability of SARS-CoV-2 to enter and exit without triggering fever and malaise, the asymptomatic period during which the majority of infections occur. Two of the virus’s most immune-suppressive genes, Orf9b, and Orf6 are over-expressed in human lung epithelial cells infected by the Alpha variant. This is no small effect. The concentration of Orf9b messenger RNA is 65 to 80 times more abundant than that produced by two early SARS-CoV-2 isolates.
Thorne et al.’s observations add to our understanding of the increased transmission of the Alpha variant. People infected with Alpha may shed more infectious viruses for a longer time before falling ill themselves. In addition to spike protein changes, more effective suppression of innate immunity may account for the longer latent period and tenfold increase in viral concentrations characteristic of Alpha infections.
This study adds two additional variables—increased potency of suppression of innate immunity and preferential viral gene expression—to our understanding of SARS-CoV-2 variants. As the authors write, “We conclude that B.1.1.7 (Alpha) has evolved beyond the Spike coding region to more effectively antagonize host innate immune responses through upregulation of specific subgenomic RNA synthesis and increased protein expression of key innate immune antagonists. We propose that more effective innate immune antagonism increases the likelihood of successful B.1.1.7 (Alpha) transmission, and may increase in vivo replication and duration of infection.”
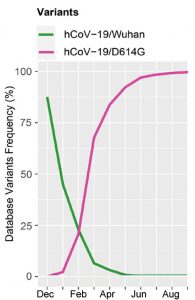
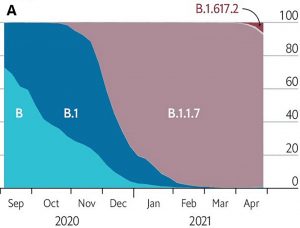
The Alpha variant first appeared in Kent, England, and within a few months displaced the predominant G clade as the most frequent source of infection. The G clade itself had previously similarly replaced the original Wuhan strain (Figure 1).
Epidemiological studies estimate that Alpha is 40-80% more infectious than the G clade viruses isolated in the early months of the pandemic. The concentration of viral RNA in patient fluids is ten times elevated and the asymptomatic period is longer. Moreover, almost twice as many of those infected require hospitalization as those infected with the earlier variants. Although the fraction of people younger than 65 infected with Alpha is higher, that observation is complicated by vaccination of the elderly and variable lockdown conditions (Figure 2).
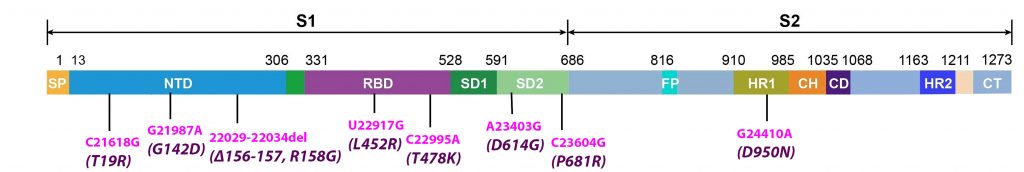
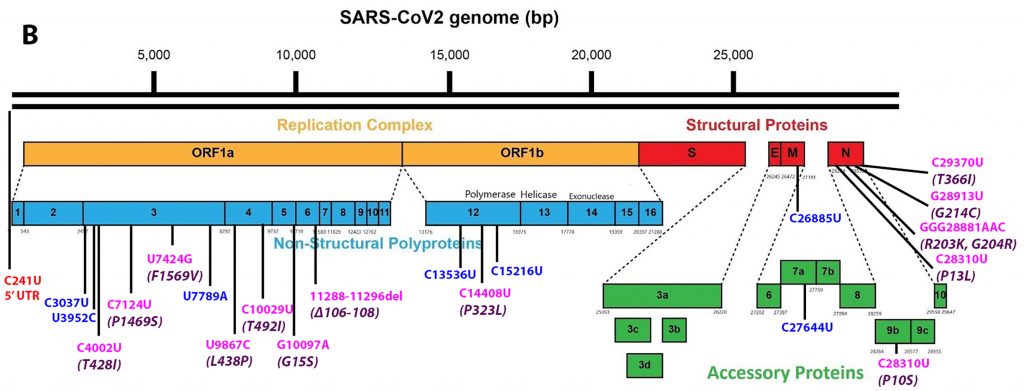
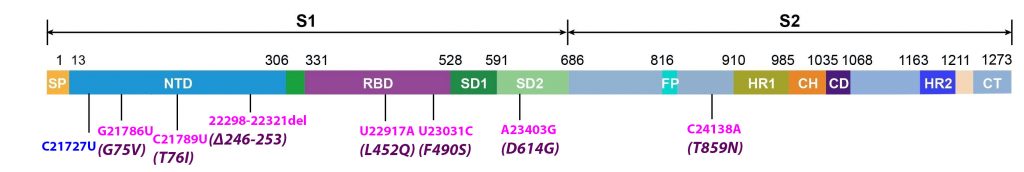
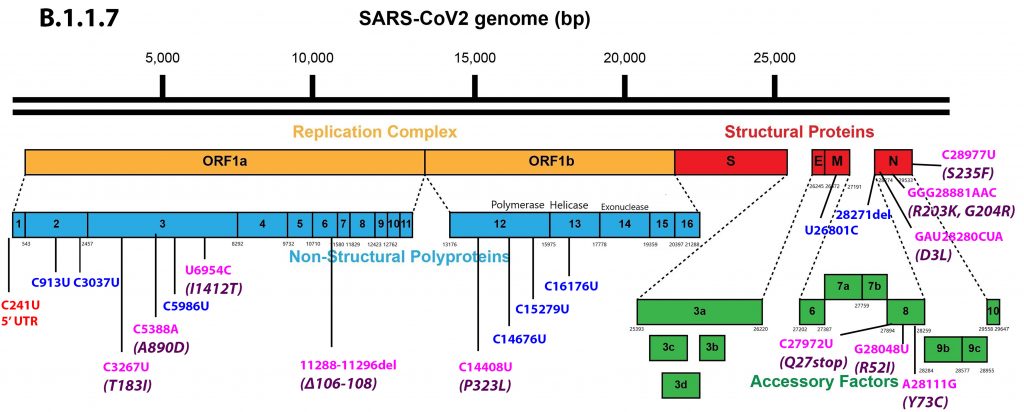
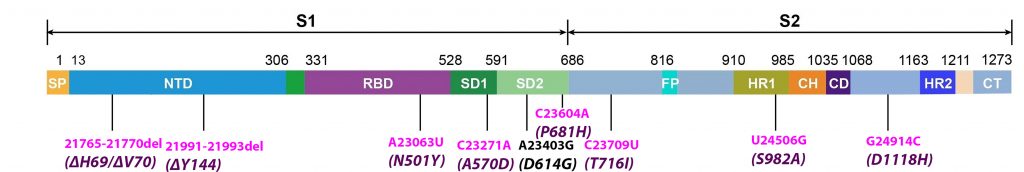
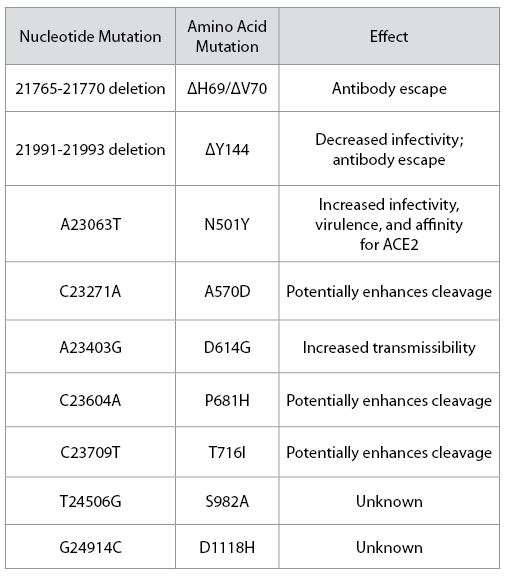
Alpha differs from the G clade B.1 virus by 48 mutations, 19 deletions, 29 single nucleotide changes, and 7 silent third codon base changes, and 22 of which result in mutations in the NSP1, 3, 6, and 12, S, Orf8, and N. This is in addition to the four mutations that distinguish the G clade virus from the consensus Wuhan genome (Figure 3).
They report the unexpected observation that the concentration of Orf9b mRNA is elevated 65 to 80 fold in cells infected with the Alpha variant as normalized to viral genomic RNA. They also observe modest increases in the mRNAs of the messenger RNAs of Orf6 and several other Orf proteins. They find a 6.5-fold increase in the Orf9b protein and a modest 1.5-2 fold increase in the Orf6 and N proteins in cells infected with the Alpha variant.
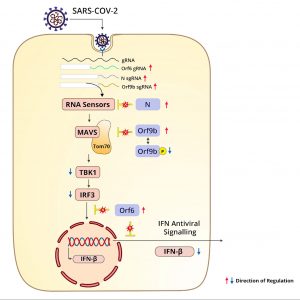
Orf9b inhibits interferon production. The authors show Orf9b protein binds to the mitochondrial antiviral signaling protein TOM70, a mitochondrial protein, to prevent its binding to the mitochondrial antiviral signaling protein (MAVS). MAVS activates Tank Binding Kinase 1 (TBK1) and IRF3. Both are required for induction of interferon in the infected cell. (Figure 5).
Thorne et al. also note a modest increase in the abundance of the nucleocapsid protein N. One explanation is that the N and Orf9b proteins are translated from the same polycistronic mRNA. In addition to packaging the full-length genome into the mature virus particle, N also acts to suppress the innate immune response (Figure 6).
Previous studies by these authors and others find that infection by early SARS-CoV-2 isolates suppresses activation of the innate immune response at early times post-infection. Here the authors show that this immune expression is more profound in cells infected by the Alpha variant. To begin with, Alpha increases suppression of interferon-beta RNA, as well as that of the intracellular concentration and secretion of interferon-beta as compared to that of earlier isolates. At early times post-infection, the Alpha variant also increases the level of suppression of a wide array of interferon-stimulated genes, including IGIT2, IFI T1 (interferon induced protein with tetratricopeptide repeats 1) RSAO2 (regional oxygen saturation), MX1 (MX dynamin like GTPase 1), and CXCL10 (C-X-C motif chemokine ligand 10). The direct and indirect effects on interferon help to explain increased resistance of the Alpha variant of cells treated with exogenous interferon-beta before and after infection.
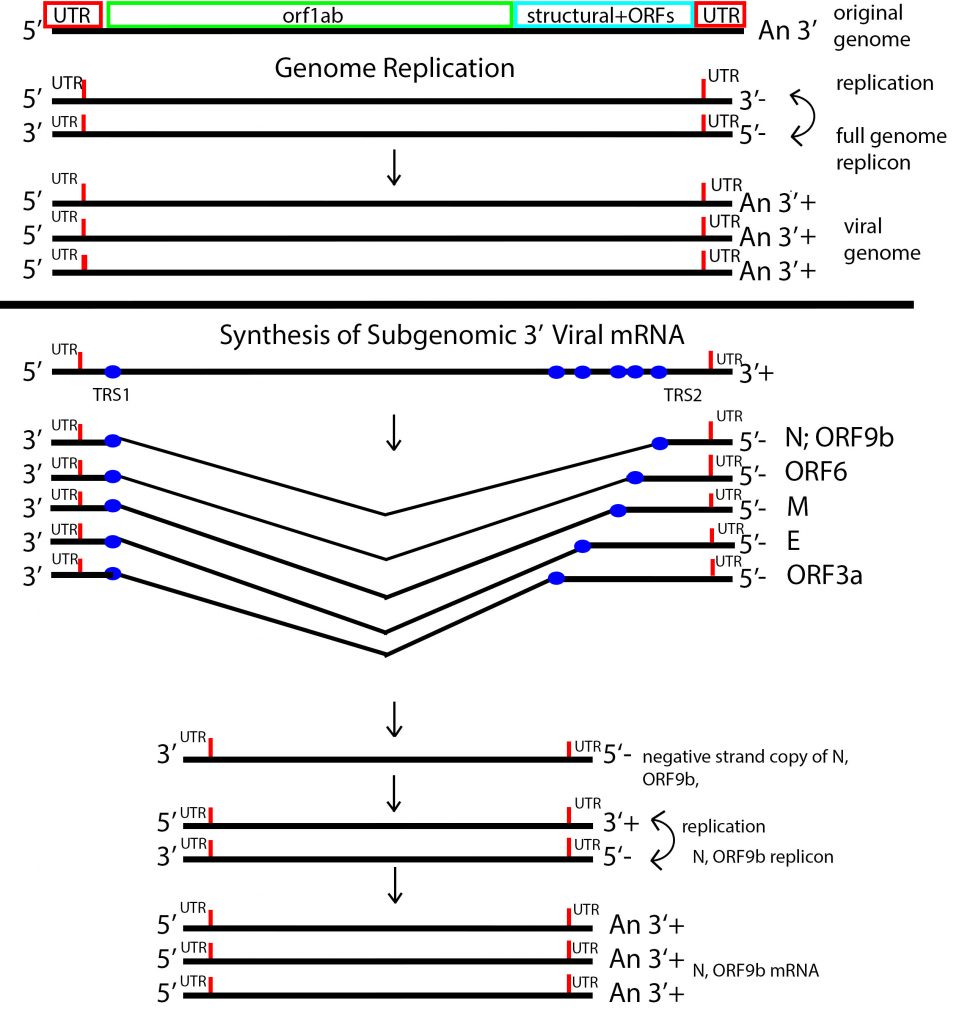
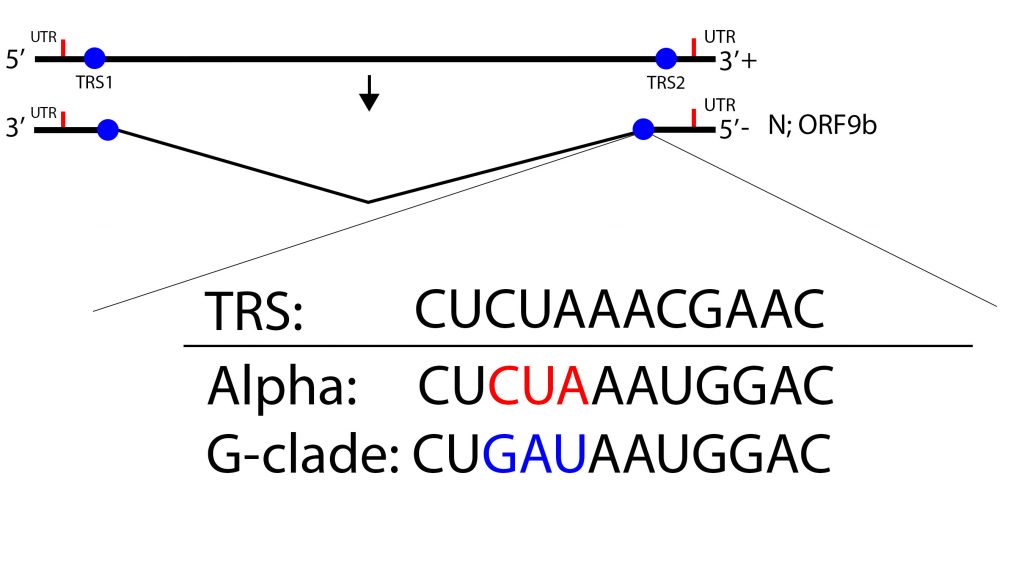
The authors provide two non-exclusive explanations of preferential expression of the N/Orf9 messenger RNA. Or9b and N occupy the same 3 prime terminal messenger RNA, albeit in different reading frames. The Alpha variant carries a six nucleotide mutation in the amino-terminal coding region of the N protein introducing a single amino acid change, N-D6L. The effect of this change on N protein activity is speculative. However, the six nucleotides mutations bring the transcription regulatory sequence (TRS) of the N/Orf9b messenger RNA closer to the consensus TRS (Figure 7). The change may increase the frequency of production of the subgenomic N/Orf9b RNAs. Indeed an excess of the N Orf/9bb and Orf6 subgenomic RNAs relative to the total RNA is observed for infections with the Alpha variant in patients infected with Alpha reference 40. It is also possible that the six-nucleotide mutation also increases the translation efficiency of both N and Orf9b. The authors do not explain the observed expression of the Orf6 mRNA (Figure 7).
Preferential mRNA expression may reflect selection for amplification of independent subgenomic replicons, each encoding separate regulatory proteins. Selection may occur at the level of cis-acting genomic sequences, such as mutations that affect the TRS sequences as proposed by Thorne et al. Preferential subgenomic replicon amplification may also be influenced by mutations that affect viral protein-coding sequences as well. For example, selective subgenomic replicon amplification occurs in influenza. Preferential amplification of defective interfering replicon results from a single amino acid change in the NS2 influenza protein.
We are still in the early days of SARS-CoV-2 evolution to increasing transmissibility and immune evasion. If our collective experience with the cold-causing coronaviruses and influenza is any guide, we have much more to learn. New variants are already emerging that are proving just how little we know about what this virus is capable of. The Delta variant, which is quickly becoming the dominant strain in all parts of the world, is more than 50% more transmissible than Alpha and almost twice as infectious as the original strain.
A Scottish study also suggests this variant is more virulent, with those infected nearly twice as likely to end up in hospital. Delta has already evolved to Delta Plus, with an additional K417N mutation which may make it even more transmissible and more capable of immune escape. And now Lambda, circulating rapidly across parts of South America, has emerged with novel mutations in the receptor binding domain that may make it more transmissible and increase the odds of reinfection (Figure 8).